Moderna Spikevax-FDA post mortem
Moderna Spikevax-FDA post mortem
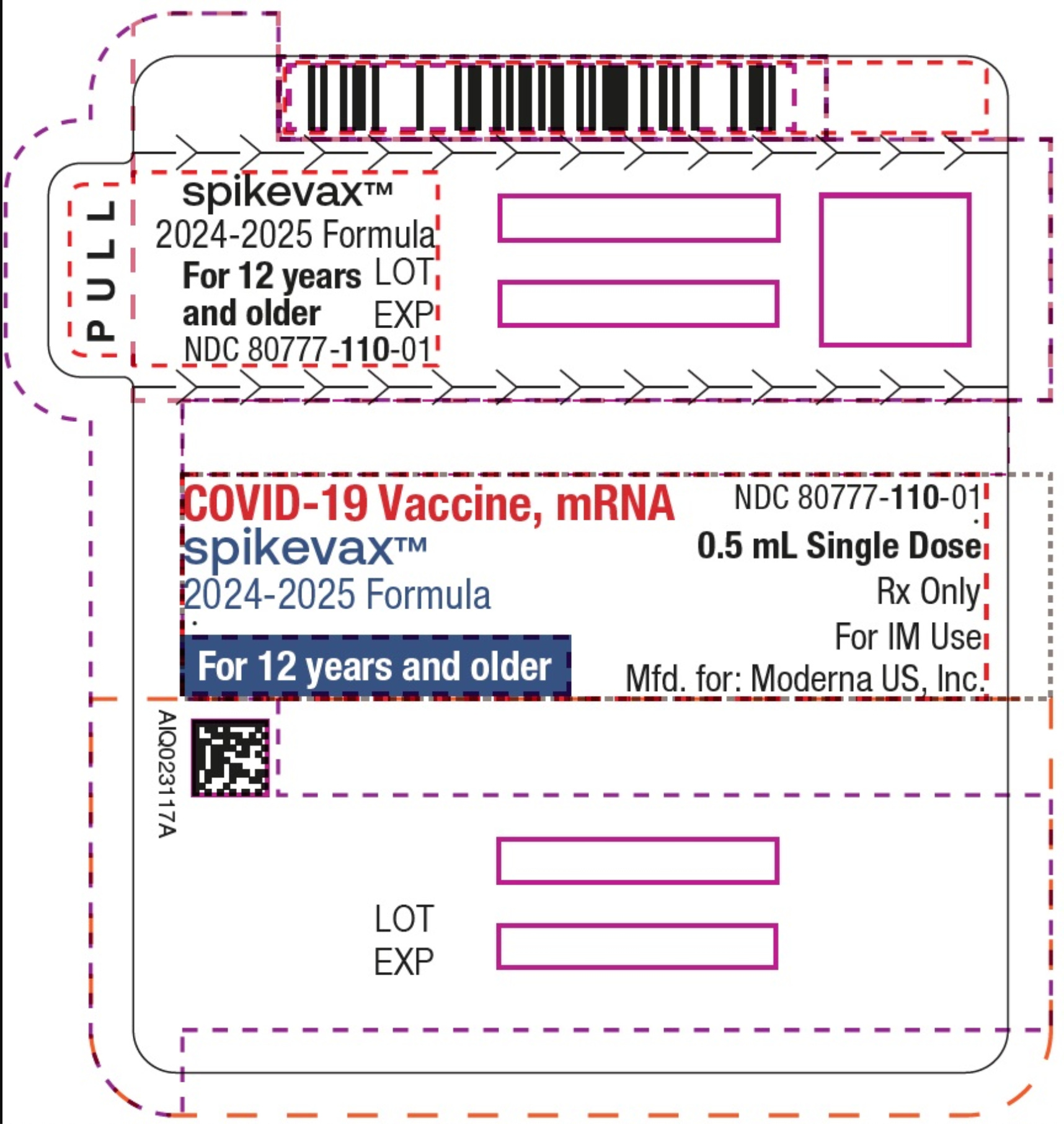
Etichettatura obsoleta per il prodotto aggiornato Moderna Spikevax 24-25. La nuova etichetta approvata dalla FDA indicherà "per i pazienti dai 6 mesi agli 11 anni a maggior rischio di sviluppare COVID-19 grave ".
Cosa è successo il 10 luglio?
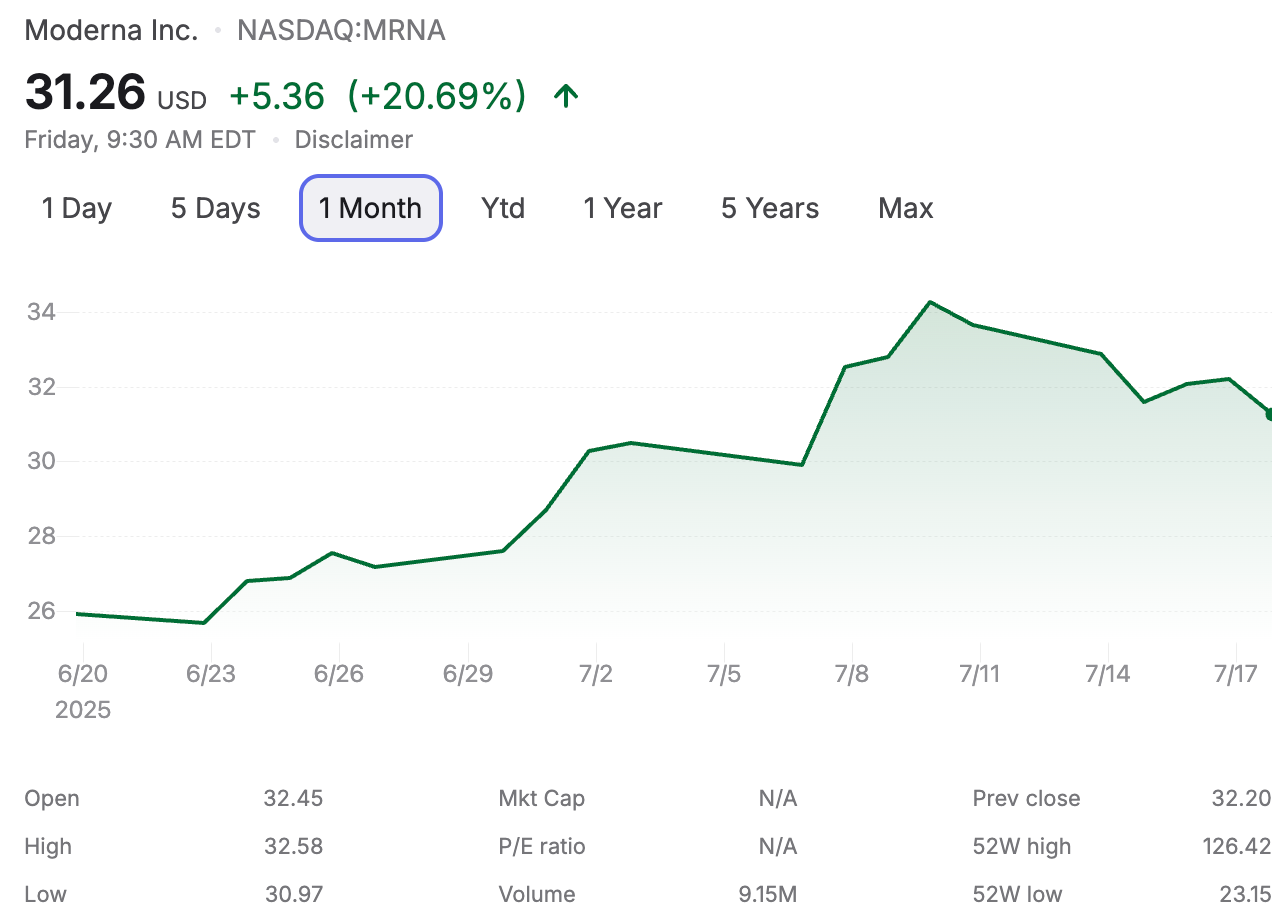
Ho ricevuto numerose richieste di chiarimento riguardo all'esito della recente autorizzazione all'immissione in commercio generale (non EUA) della FDA del 10 luglio 2025 per il vaccino SpikeVax di Moderna per uso pediatrico. La verità è che non so esattamente cosa sia successo alla FDA per arrivare a questo risultato. Come molti, sono rimasto scioccato e deluso. Moderna era felicissima. Il valore delle loro azioni e la loro capitalizzazione di mercato sono aumentati vertiginosamente, con un'impennata iniziata quattro giorni prima dell'annuncio, suggerendo che qualcuno avesse informazioni privilegiate. Se fosse vero, si tratterebbe probabilmente di insider trading. Questo sarebbe esattamente il tipo di violazione delle regole che molti si aspettano da questa azienda e dal settore. Ai fini di questa discussione, potete considerarla un'ipotesi.
Il vaccino Spikevax COVID-19 di Moderna ha ricevuto la piena approvazione della FDA statunitense per i bambini di età compresa tra 6 mesi e 11 anni a maggior rischio di sviluppare la malattia grave da COVID-19 , come annunciato il 10 luglio 2025. Questa approvazione sostituisce la precedente disponibilità del vaccino in base all'autorizzazione all'uso di emergenza (EUA) per questa fascia d'età. Spikevax era stato precedentemente approvato per gli adulti di età pari o superiore a 65 anni e per gli individui di età compresa tra 6 mesi e 64 anni a maggior rischio di sviluppare la malattia grave.
Spikevax di Moderna ha ottenuto la prima approvazione della FDA nel gennaio 2022 per uso adulto . Da allora, il vaccino è stato aggiornato, incluso il targeting per la variante KP.2 del SARS-CoV-2, ed è stato esteso per coprire fasce d'età più ampie. Moderna prevede di rendere disponibile la versione aggiornata del vaccino Spikevax per le popolazioni idonee negli Stati Uniti prima della stagione virale respiratoria 2025-2026.
L'approvazione si è basata sulla revisione scientifica della FDA, con l'amministratore delegato di Moderna, Stéphane Bancel, che ha sottolineato la continua minaccia del COVID-19 per i bambini, in particolare quelli con patologie preesistenti. Nonostante l'approvazione, i critici hanno sollevato preoccupazioni circa la mancanza di dati sulla sicurezza pediatrica e il potenziale rischio di gravi eventi avversi, tra cui miocardite e pericardite. Il foglietto illustrativo di Spikevax elenca diversi eventi avversi, come difficoltà respiratorie, gonfiore del viso e della gola, tachicardia, vertigini e debolezza.
La FDA e Moderna hanno riconosciuto questi rischi e i foglietti illustrativi aggiornati ora includono avvertenze più severe su miocardite e pericardite. Il vaccino è destinato a individui a maggior rischio di sviluppare la malattia in forma grave e la vaccinazione potrebbe non proteggere tutti i soggetti che lo ricevono.
Moderna ha anche altri vaccini nel suo portfolio, tra cui mNEXSPIKE (mRNA-1283), approvato a giugno 2025 per gli adulti di età pari o superiore a 65 anni e per i soggetti di età compresa tra 12 e 64 anni con almeno un fattore di rischio per la malattia grave. Questa approvazione si basa sui dati dello studio di fase III NextCOVE, che ha dimostrato la non inferiorità di mNEXSPIKE rispetto a Spikevax.
Ci sono diverse questioni irrisolte legate a questa azione.
Perché approvare Spikevax di Moderna per questa indicazione e non concedere un'approvazione analoga per il prodotto di Pfizer?
Il commissario della FDA, il dott. Marty Makary, era a conoscenza di questa decisione e l'ha approvata, oppure è stata decisa e attuata a livello del direttore del Centro per la valutazione e la ricerca biologica (CBER) della FDA, Vinay Prasad?
È stata una coincidenza il fatto che questa decisione sia stata presa mentre il Segretario Kennedy e il suo vice capo di gabinetto Stephanie Spear erano in vacanza?
Questa decisione è stata presa nel contesto di un piano strategico più ampio all'interno dell'HHS?
Quali sono le condizioni preesistenti che la FDA considera " a maggior rischio di sviluppare la malattia grave da COVID-19 nei bambini dai 6 mesi agli 11 anni"? In altre parole, per usare un linguaggio semplice, quali sono le specifiche patologie infantili che potrebbero affliggere tuo figlio e che lo renderebbero idoneo a questo prodotto autorizzato dalla FDA?
Quali e dove sono i dati che dimostrano che i benefici derivanti dalla somministrazione di questo prodotto superano i rischi per i bambini affetti da queste specifiche patologie? I medici e i genitori potranno visionare i dati dell'analisi rischio/beneficio, che (presumibilmente) la FDA ha utilizzato per giustificare questa decisione?
Qual è il numero di " bambini di età compresa tra 6 mesi e 11 anni a maggior rischio di COVID-19 grave" che devono essere trattati con questo prodotto per prevenire inequivocabilmente un decesso o un ricovero ospedaliero evitabile a causa di COVID-19 grave? Ci sarà consentito di vedere questo calcolo per ciascuna delle condizioni preesistenti che la FDA ritiene associate a un " maggiore rischio di COVID-19 grave"?
Questi sono i dati di cui pediatri e genitori hanno bisogno per prendere decisioni consapevoli sull'accettazione di questo prodotto per i propri figli. Affermazioni vaghe sull'aumento del rischio non saranno sufficienti. I genitori devono sapere di quali specifiche condizioni preesistenti devono preoccuparsi e quanto sia significativo il rischio di contrarre una forma grave di COVID-19 per il proprio figlio affetto da tale condizione.
I giorni in cui l'opinione pubblica tollerava l'atteggiamento paternalistico della FDA, del tipo "noi siamo i professionisti e sappiamo tutto, quindi non avete bisogno di vedere i dati e le nostre analisi", sono finiti.
Mostrateci i dati. Chiaro e semplice.
Ma chi parlerà per i danneggiati? Chi proteggerà i genitori e i loro figli? Per essere schietti, la FDA/CBER ha concesso un'autorizzazione all'immissione in commercio generale limitata per questo prodotto, ma chi deciderà come utilizzarlo? Naturalmente, in un mondo razionale, i singoli pediatri, i medici di base e i genitori che rilasciano il consenso informato sono in prima linea in questa decisione.
Ma la verità è che la pratica medica negli Stati Uniti è in ultima analisi determinata dalla responsabilità legale. Le preoccupazioni relative alla responsabilità legale guidano tutto, dallo studio medico fino agli ospedali e ai sistemi "sanitari", fino alle compagnie assicurative. E la responsabilità legale dipende dal rispetto o meno degli "standard di cura" in un singolo caso. Nel caso dei produttori di vaccini, hanno esercitato con successo pressioni sul governo per ottenere una copertura completa della responsabilità, quindi in un certo senso non hanno alcun interesse in questa lotta, se non per il fatto che lo standard di cura determina quante dosi di prodotto possono vendere.
Chi stabilirà lo standard di cura in relazione a questo prodotto?
Storicamente, la risposta è stata il Comitato Consultivo sulle Pratiche di Immunizzazione (ACIP) del CDC. Questo semplice fatto spiega perché i cagnolini dei media aziendali del settore farmaceutico abbiano urlato alla luna per la decisione del Segretario Kennedy di ritirare il vecchio ACIP, che si era trasformato in un semplice timbro per la FDA e il settore farmaceutico, e di sostituirlo con membri che insistessero nel valutare in modo indipendente i dati effettivi a supporto (o meno) dell'uso di un vaccino contro le malattie infettive o di prodotti correlati per le indicazioni autorizzate.
Riesci a capirlo ? Mi riferisco all'uso del termine coniato da Robert Heinlein nel suo immortale "Straniero in terra straniera", che significa comprendere qualcosa in profondità a livello emotivo, intellettuale e sociale.
Tecnicamente, secondo la Costituzione, FDA/CBER autorizza il trasporto e la commercializzazione interstatale di prodotti e dispositivi medici. Il CDC/ACIP fornisce raccomandazioni al Direttore del CDC (e al Segretario HHS) sugli standard di cura per i vaccini contro le malattie infettive e i prodotti correlati. Le raccomandazioni dell'ACIP diventano raccomandazioni ufficiali del CDC solo dopo essere state accettate dal Direttore del CDC. Il Direttore del CDC può, e ha già fatto, annullare le raccomandazioni consultive dell'ACIP in rarissime occasioni.
Per statuto, l'ACIP è tenuto a discutere la questione di quale consiglio fornire al Direttore del CDC alla prossima riunione dell'ACIP, in seguito all'autorizzazione da parte della FDA di un nuovo vaccino. L'ACIP non ha soddisfatto questo requisito nel caso del prodotto Moderna mNEXSPIKE , recentemente autorizzato dalla FDA . Non ho idea se e quando l'ACIP potrà affrontare questa azione della FDA.
Per quanto riguarda la recente autorizzazione all'immissione in commercio da parte della FDA per Spikevax, cosa farà ora l'ACIP? Purtroppo, in quanto membro dell'ACIP (siamo tutti "volontari", tra l'altro), mi è vietato commentare i futuri piani dell'ACIP al di là di quanto sia di pubblico dominio. Il che è giusto che sia. Non parlo, e non posso farlo, a nome dell'ACIP, del CDC o del governo degli Stati Uniti, e le deliberazioni e i piani interni dell'ACIP sono considerati informazioni riservate.
Tutto ciò significa che il direttore della FDA/CBER, Vinay Prasad, ha appena gettato una patata bollente direttamente in grembo all'ACIP, che ora si frappone tra questa decisione della FDA e milioni di genitori statunitensi e i loro figli. Beh, tecnicamente, l'ACIP non fa altro che consigliare il direttore del CDC, attualmente in attesa dell'approvazione del Senato. E sapete cosa significa. Il Dott. Prasad ha anche, di fatto, gettato la stessa patata bollente negli ingranaggi del processo di conferma del Senato.
A titolo di chiarimento e contesto, Moderna giustifica l'introduzione e la commercializzazione del prodotto mNEXSPIKE in parallelo al suo prodotto Spikevax sostenendo che offre vantaggi di progettazione che forniscono protezione con un quinto della dose di Spikevax e consentono la conservazione tra 2 °C e 8 °C fino a 90 giorni. Spikevax può essere conservato a queste temperature fino a 60 giorni.
Il nuovo vaccino può essere conservato a temperature comprese tra 8°C e 25°C per un tempo doppio rispetto a Spikevax. I miglioramenti derivano da una differenza nella progettazione dei vaccini. Spikevax codifica una versione modificata della proteina Spike virale a lunghezza intera, che ha dimostrato di essere associata a un'ampia gamma di tossicità. Il nuovo vaccino codifica il dominio di legame al recettore e il dominio N-terminale del virus. Una lunghezza inferiore dell'mRNA è associata a una maggiore stabilità. Presumibilmente, questa versione redatta tramite ricombinazione della proteina Spike a lunghezza intera sarà anche priva delle note tossicità associate sia alla proteina Spike codificata da virus, sia alle proteine Spike modificate utilizzate in praticamente tutti gli altri prodotti "vaccinabili" contro il COVID-19 autorizzati dalla FDA.
Chi lo sapeva, cosa e quando, e c'erano considerazioni strategiche e tattiche più ampie?
Ho una conoscenza limitata di queste questioni. La triste verità è che questa decisione sull'autorizzazione FDA/CBER per Spikevax " per bambini dai 6 mesi agli 11 anni a maggior rischio di COVID-19 grave " è stata resa pubblica da Moderna mentre sia il Segretario dell'HHS (JFKjr) che la sua fidata vice capo di gabinetto Stefanie Spears erano in vacanza. E Moderna ha certamente fatto il possibile per far fruttare le sue energie finché il sole splendeva su di loro.
Né il Segretario HHS né il suo vice Capo di Gabinetto (dCOS) sono stati informati o informati di questa decisione. Il Commissario della FDA era a conoscenza? L'ex Capo di Gabinetto HHS Heather Flick lo era? Il Presidente degli Stati Uniti o il suo Capo di Gabinetto (COS) lo erano? Non conosco le risposte a queste domande. Quello che so è che poco dopo il ritorno dalle vacanze del Segretario HHS e del suo dCOS, è avvenuta un'importante riorganizzazione della dirigenza HHS. E che il Presidente degli Stati Uniti e il suo COS sono stati informati di questa decisione. Ora c'è un nuovo Capo di Gabinetto HHS. Il dCOS per le politiche è stato portato via dai locali ed era così sconvolto che ha schiantato la sua auto contro il veicolo fornito dal governo al Segretario. E sento rulli di tamburo in lontananza che segnalano che gli indigeni credono che saranno imminenti ulteriori cambiamenti organizzativi.
Per coloro, me compreso, che hanno insistito sul fatto che ci sarebbero state conseguenze per non aver informato la catena di comando in merito a questa deludente e critica decisione del direttore FDA/CBER, c'è conforto nel sapere che ci sono state conseguenze immediate. Che siate d'accordo o meno con la decisione, spero che tutti comprendano che la burocrazia federale deve riconoscere che ci saranno conseguenze per non aver informato la propria catena di comando su decisioni politicamente delicate. Una scossa breve e intensa era esattamente ciò che il medico aveva raccomandato.
Per quanto riguarda strategia e tattica
La narrazione su Twitter di MAHA, dopo l'approvazione da parte della FDA del vaccino SPIKEVAX di Moderna per i bambini ad alto rischio, è che si sia trattato di un tradimento da parte del Segretario Kennedy. Ma questo è l'OPPOSTO di ciò che è accaduto. Il Segretario era in vacanza e non è stato nemmeno informato della decisione. Era una vacanza lavorativa, e ho la conferma personale che ha ricevuto molte chiamate, briefing e ha gestito costantemente decisioni strategiche e tattiche durante la sua meritata e necessaria vacanza. Lui (e la sua amata moglie) non si sono concessi una vacanza da quando hanno lanciato la campagna presidenziale.
La realtà è che il direttore del CBER, Vinay Prasad, ha scavalcato le raccomandazioni degli enti regolatori della FDA, che avevano raccomandato l'approvazione di tutti e tre i vaccini COVID attualmente in fase di EUA per tutte le fasce d'età, compresi i bambini sani . Il che solleva la domanda: perché abbiamo ancora un'autorizzazione all'uso di emergenza per questi prodotti quando chiaramente non esiste alcuna emergenza medica?
La giustificazione di Prasad per questo rifiuto dovrebbe rassicurare l'intero movimento per la libertà medica. Nella sua lettera di giustificazione, ha affermato:
Esiste una sostanziale certezza di un beneficio clinico netto derivante dalla vaccinazione di bambini sani con questo vaccino a mRNA? La risposta dell'Ufficio Comunicazione, Sensibilizzazione e Sviluppo (OCD) del Centro per la Valutazione e la Ricerca sui Prodotti Biologici (CBER) è, al momento, no, in base alle migliori informazioni disponibili.
Moderna non ha mai dimostrato una riduzione dei casi gravi di COVID-19, dei ricoveri ospedalieri, dei ricoveri in terapia intensiva o dei decessi in uno studio randomizzato sui bambini.
Moderna non ha dimostrato che la vaccinazione contro il COVID-19 riduca la durata del COVID-19 o la trasmissione in alcun contesto e a nessuna età, con dati di alta qualità. Né il richiedente né una terza parte hanno dimostrato una riduzione dei giorni di scuola persi con dati di alta qualità.
La vaccinazione di questi individui (bambini sani dotati di immunità naturale) comporta un'enorme incertezza circa il fatto che i benefici superino i rischi.
Sebbene i vaccini contro il COVID-19 siano stati somministrati a miliardi di persone e i danni siano stati studiati a fondo, nessuno sa se questi prodotti abbiano effetti dannosi che si manifestino solo 10 o 20 anni dopo, poiché tale limite temporale è necessario. È ignorante affermare che non siano possibili rischi a lungo termine sconosciuti.
Gli anticorpi non sono il gold standard della scienza e non si può essere certi di un beneficio clinico netto semplicemente perché gli anticorpi aumentano. Le dosi di vaccino possono aumentare gli anticorpi, ma non riescono a migliorare ulteriormente i risultati clinici.
Saranno necessari studi clinici randomizzati per approvare questi prodotti per individui sani.
La FDA è in ultima analisi responsabile nei confronti del popolo americano, e gli americani hanno dichiarato in modo schiacciante di ritenere che le prove a sostegno della vaccinazione di un bambino sano con un prodotto a mRNA per il COVID-19 non siano sufficienti a spingerli ad agire. Il CBER OCD, dopo un attento esame delle prove scientifiche, concorda con la stragrande maggioranza degli americani.
Personalmente, in qualità di medico esperto in questo campo, sono d'accordo con tutti questi punti.
E questo arriva subito dopo che il Commissario della FDA Makary ha dichiarato a Epoch Times di "conoscere personalmente persone che sono state danneggiate dal vaccino. Conosco personalmente amici che hanno perso una persona cara a causa del vaccino mRNA contro il COVID".
Ha risposto ai critici che affermano che lui e Kennedy non stanno facendo abbastanza per i vaccini contro il COVID-19: "Quindi le persone hanno il diritto di essere arrabbiate. Sono state ingannate su diversi aspetti della pandemia di COVID. È stato loro ordinato di mettersi in fila per la vaccinazione anche se erano sane, a basso rischio e avevano già anticorpi circolanti. Le persone hanno il diritto di essere arrabbiate, ma vorrei chiedere loro di essere pazienti con noi mentre lo facciamo nel modo scientificamente corretto".
Le cose si stanno muovendo nella giusta direzione. Il Segretario Kennedy ha sempre affermato che dobbiamo restituire la scienza d'oro all'HHS. Quando i dati saranno disponibili e le raccomandazioni saranno formulate, ho piena fiducia che si muoverà di conseguenza.
Pensate che qualsiasi FDA precedente avrebbe rifiutato l'approvazione di qualsiasi vaccino, per non parlare di quello contro il COVID-19? Le prove suggeriscono il contrario. Questo è un enorme passo avanti.
Non ho alcun impatto o controllo sulle decisioni prese dalla FDA. Hanno preso una decisione, nel bene e nel male. La decisione è stata presa senza il consenso o la consapevolezza del Segretario dell'HHS.
Lo statuto dell'ACIP stabilisce che spetta ora all'ACIP raccomandare al Direttore del CDC le linee guida per gli standard di cura per l'uso di questo prodotto. E spetterà al Direttore del CDC accettare o respingere tali raccomandazioni. Questi sono fatti pubblici, quindi sono libero di menzionarli. Il tempo ci dirà come l'ACIP assolverà a questa responsabilità.
E no, nonostante quanto affermato da alcuni agenti del caos divisivo, fino a oggi il nuovo ACIP non ha votato per supportare o meno NESSUNA raccomandazione sui prodotti vaccinali contro il COVID-19.
Per chi fosse interessato ad approfondire l'argomento, consiglio la seguente pre-print. Il primo autore di questo lavoro è un collega membro dell'ACIP e Professore Ordinario del MIT per il quale nutro profondo rispetto. L'autore principale è il Surgeon General della Florida, per il quale nutro anch'egli profondo rispetto. mRNA-1273 = Spikevax, e questi dati supportano il fatto che, se il Direttore del CBER Prasad riteneva necessario approvare almeno un prodotto a base di mRNA per il COVID-19 per proteggere i bambini ad alto rischio di genitori che richiedono l'accesso a un prodotto di richiamo a base di mRNA per il COVID-19 per il proprio figlio, Spikevax era una scelta ragionevole. Era lui a dover prendere questa decisione, e l'ha presa, nel bene e nel male.
In una società normale e giusta, questo manoscritto avrebbe già completato la revisione paritaria e sarebbe stato pubblicato su una rivista di ricerca medica di alto profilo. Il fatto che ciò non sia avvenuto dimostra che la propaganda, la censura e la guerra psicologica sui prodotti mRNA per il COVID-19 continuano.
Mortalità per tutte le cause a dodici mesi dopo la vaccinazione iniziale contro il COVID-19 con Pfizer-BioNTech o mRNA-1273 tra gli adulti residenti in Florida
Retsef Levi , Fahad Mansuri, Melissa M. Jordan, Joseph A. Ladapo
ASTRATTO
Obiettivo Esaminare l'impatto relativo della serie iniziale di RNA messaggero (mRNA) BNT162b2 (Pfizer) e mRNA-1273 (Moderna) sulla mortalità per tutte le cause e non COVID-19 tra i residenti della Florida.
Progettazione Coorte abbinata con valutazioni cumulative e aggiustate del rischio durante un follow-up di 12 mesi.
Creazione di database sanitari pubblici a livello statale della Florida con dati sulla vaccinazione contro il COVID-19, caratteristiche sociodemografiche dei destinatari del vaccino, luogo di vaccinazione e statistiche vitali.
Partecipanti Coorte abbinata di 1.470.100 adulti residenti in Florida non istituzionalizzati che hanno ricevuto almeno due dosi, a meno di sei settimane di distanza, del vaccino BNT162b2 o mRNA-1273 mRNA tra il 18 dicembre 2020 e il 31 agosto 2021.
Intervento Vaccinazione iniziale con due dosi di BNT162b2 o mRNA-1273
Principali misure di esito Mortalità per tutte le cause, cardiovascolare, COVID-19 e non COVID-19 entro 12 mesi dalla seconda dose del vaccino COVID-19
Risultati : 9.162.484 adulti residenti in Florida non istituzionalizzati soddisfacevano i criteri di inclusione, inclusi 5.328.226 destinatari del vaccino BNT162b2 e 3.834.258 destinatari del vaccino mRNA-1273. Un totale di 1.470.100 vaccinati sono stati abbinati 1 a 1 in base a sette criteri, tra cui l'area censuaria. Rispetto ai destinatari di mRNA-1273, i destinatari di BNT162b2 presentavano un rischio significativamente più elevato di mortalità per tutte le cause (847,2 contro 617,9 decessi ogni 100.000; odds ratio, OR [95% CI]: 1,384 [1,331, 1,439]), mortalità cardiovascolare (248,7 contro 162,4 decessi ogni 100.000 persone; OR [95% CI]: 1,540 [1,431, 1,657]), mortalità COVID-19 (55,5 contro 29,5 decessi ogni 100.000 persone; OR [95% CI]: 1,882 [1,596, 2,220]) e mortalità non COVID-19 (791,6 contro 588,4 decessi ogni 100.000 persone; OR [95% CI]: 1,356 [1,303, 1,412]). I risultati del controllo negativo non hanno mostrato alcuna indicazione di significativi fattori di confondimento residui non osservati.
Conclusione Gli adulti della Florida che hanno ricevuto BNT162b2 hanno mostrato un rischio significativamente più elevato di mortalità a 12 mesi per tutte le cause, malattie cardiovascolari, COVID-19 e non COVID-19 rispetto ai corrispondenti riceventi mRNA-1273. Questi risultati suggeriscono effetti differenziali aspecifici dei vaccini BNT162b2 e mRNA-1273 contro il COVID-19 e potenziali effetti avversi preoccupanti sulla mortalità per tutte le cause e per malattie cardiovascolari. Sottolineano la necessità di valutare i vaccini utilizzando endpoint clinici che si estendano oltre le malattie a cui sono destinati.
Cosa si sa già su questo argomento
I vaccini possono avere effetti sulla salute che vanno oltre le malattie a cui sono destinati, compresi potenziali effetti sulla mortalità per tutte le cause.
L'impatto relativo delle serie iniziali di RNA messaggero (mRNA) BNT162b2 (Pfizer) e mRNA-1273 (Moderna) sulla mortalità per tutte le cause e sulla mortalità non correlata al COVID-19 non è stato ben studiato.
Cosa aggiunge questo argomento
Gli adulti della Florida che hanno ricevuto BNT162b2 hanno avuto un rischio significativamente più elevato di mortalità a 12 mesi per tutte le cause, cardiovascolare, COVID-19 e non COVID-19 rispetto ai riceventi mRNA-1273 attentamente abbinati.
Questi risultati suggeriscono effetti differenziali non specifici dei vaccini BNT162b2 e mRNA-1273 contro il COVID-19 e potenziali effetti avversi preoccupanti sulla mortalità per tutte le cause e cardiovascolare.
Dichiarazione di interessi concorrenti
MJ, FM e JL hanno ricevuto il supporto del Dipartimento della Salute della Florida e JL è il chirurgo generale dello Stato; RL ha un rapporto finanziario con la Bluebell Foundation/Chicago Community Trust; non ci sono altre relazioni o attività che potrebbero sembrare aver influenzato il lavoro presentato.
Dichiarazione di finanziamento
Lo studio non ha ricevuto alcun finanziamento esterno.



Commenti
Posta un commento